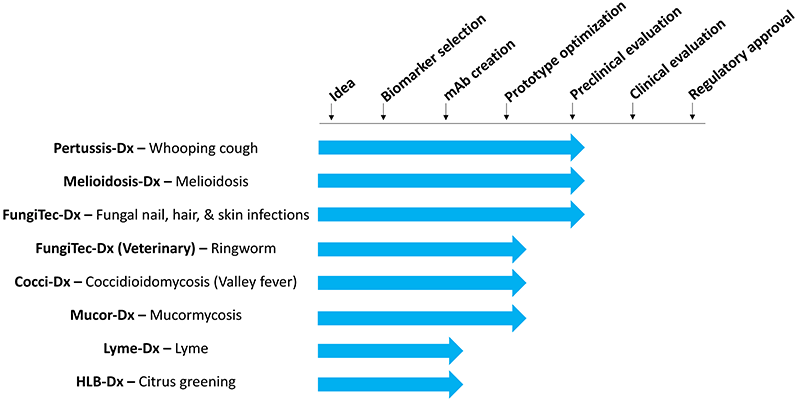
Idea – DxDiscovery builds tests on the antigen-detection immunoassay platform. This type of test detects molecules made by pathogenic microbes during disease. Detection of these microbial biomarkers tells the clinician that an active infection is present. We apply this platform to diseases where i) there is no existing diagnostic, or ii) existing diagnostics are too slow, expensive, or inaccessible due to high infrastructure requirements. Infrastructure includes expensive equipment, electricity, or specialized user expertise. Overall, DxDiscovery’s product development pipeline prioritizes diseases where clinicians require a point-of-care diagnosis (i.e. fast turn-around time, minimal infrastructure, and low cost).
Choosing an effective biomarker – In some cases, candidate microbial targets can be found by careful review of the published scientific literature or from understanding the specific disease’s pathogenesis. An example of the former are fungal mannans, which are well-established biomarkers of many invasive fungal infection, including coccidioidomycosis. An example of the latter would be leveraging the knowledge that the most abundant virulence determinants on the surface of Bordetella pertussis bacteria are highly likely to be effective and immunogenic targets.
In other cases, the pathogen is known, but candidate target biomarkers made by that pathogen are still unknown and must be discovered. An example of an infection where a set of candidate targets is not known is Huanglongbing (citrus greening disease). DxDiscovery uses a combination of hypothesis-driven and data-driven experimental approaches to discover new microbial biomarkers to target in such cases.
Monoclonal antibody creation – High-quality monoclonal antibodies (mAbs) are critical for successful immunoassay diagnostics. For each diagnostic development project, DxDiscovery produces large libraries of candidate mAbs in-house. These libraries are then carefully winnowed down to the best-performing mAbs by asking the following questions. Is the antibody sensitive enough to detect even very small amounts of the pathogen of interest? Is the antibody specific to the desired pathogen, e.g. without cross-reactivity to other microbes? Is the antibody inclusive and strongly reactive across all strains of the desired pathogen? Is the antibody easy to manufacture at scales that will enable commercial production of affordable assays (e.g. stable antibody-secreting cell line with high antibody yield per liter of cell culture)?
Immunoassay optimization – Once the best mAbs have been chosen, then immunoassays containing the mAb(s) are optimized for analytical sensitivity, specificity, and time to result. DxDiscovery typically evaluates mAbs for both antigen-capture ELISA performance and lateral flow immunoassay performance. The final assay must meet the real-world performance needs of the clinical end-user, which vary project to project. For example, affordability and accessibility might be a need best met with a visual readout, colloidal gold-based, lateral flow immunoassay for one disease. Whereas another disease might require exquisite picogram per milliliter analytical sensitivity to achieve the needed clinical sensitivity for rule-out diagnostic usage. This level of sensitivity might require a fluorescent lateral flow immunoassay reader, special specimen processing steps, and/or the ELISA format. Consequently, prototype optimization is custom-tailored to each project and is consciously done with the end-users’ needs in mind.
Regulatory approval – To achieve regulatory approval by the FDA or other international regulatory agencies, prototype immunoassays must pass a battery of pre-clinical and clinical evaluations. Preclinical evaluations focus on the analytical performance of the assay (e.g. limit of detection, precision and reproducibility, cross-reactivity, kit stability and ruggedness, etc.). Clinical evaluations compare the performance of the prototype immunoassay to an established reference method using the same set of patient specimens. The prototype assay must have a high level of agreement with the reference method to be approved for clinic use. Overall, this means that real patient specimens diagnosed as infected by the reference method must also show positive results with the prototype assay, while specimens diagnosed as negative by the reference method must also show negative results with the prototype assay.